![Overview: Chiari Malformation [Revised]](https://dev.chiaribridges.org/wp-content/uploads/2016/10/Fotolia_79774600_XS.jpg)
CHIARI MALFORMATIONS (PRONOUNCED: KEE-AH-REE) ARE STRUCTURAL DEFECTS IN WHICH THE CEREBELLUM, THE HIND PART OF THE BRAIN, DESCENDS BELOW THE FORAMEN MAGNUM INTO THE SPINAL CANAL.
While Arnold Chiari Malformation (Type 2) was first identified in the late 19th century by the Austrian pathologist Hans Chiari, much of the current medical knowledge has developed since 1985 with the expanded use of Magnetic Resonance Imaging (MRI). The number of patients diagnosed with Chiari malformations continues to increase, and with that increase Chiari Malformation is getting some of the attention the condition has always demanded.

Chiari malformations (“CMs”) are neurological disorders in which the cerebellum extends out of the skull and into the spinal canal, which in turn blocks the flow of cerebrospinal fluid, puts pressure on the brainstem and spine, and may result in varying degrees of nerve compression. Once thought to occur in 1 in 1000 people, it is now believed to be much more frequent of an occurrence. A 2016 pediatric study found it to occur in 1 in 100 children[1]. However, since the most common type (Type 1) tends to become symptomatic during late teens and early adulthood, it is likely to be much more common when adults are factored in. Females are more likely than males to have a Chiari Malformation (at a ratio of 3:1), and significantly higher amongst those with both Chiari Malformation and Ehlers-Danlos Syndrome (9:1)[2]. We affectionately refer to those that live with this condition, including the attendant pain and frequent disregard from the medical community, as Chiarians (regardless of whether they have had surgical intervention or not).
While some Chiarians are symptomatic throughout their lifetime, the vast majority of Chiarians (those with Type 1) develop symptoms in their late teens or early adulthood. Those symptoms can range from mild to crippling, and can become severe enough to cause paralysis (often associated with syringomyelia) or death.
WHAT CAUSES A CHIARI MALFORMATION?
Multiple factors have been identified which can either cause or attribute to Chiari malformations. Although they too were once thought to be rare, Acquired Chiari malformations are now being diagnosed in increasing numbers. A brief overview of what each of these labels entail, together with a summary of the different types of CM’s, is provided below:
- Congenital Chiari, is believed to be caused by a posterior cranial fossa hypoplasia (PCFH)[3], which can also be caused by a connective tissue disorder such as Ehlers-Danlos.[4] While the cerebellum continued to grow in utero, the posterior skull failed to grow proportionately. Problems resulting from this size discrepancy continue and eventually the overcrowding of the hindbrain squeezes the cerebral tonsils downward into the opening of the spinal canal (cranial constriction). While the herniation of the cerebellar tonsil(s) can take place during gestation or after birth, because the cause is 100% congenital, and the process most likely began in utero, it is usually considered a Congenital Chiari Malformation when the only pathology found is a small posterior cranial fossa. In one large study, they found those with a Chiari Malformation and no associated etiological/pathological co-factors, with only slightly over 52% having a small PCF. When other co-factors were present, the number of Chiarians found with a small PCF plummeted, and therefore it should be considered acquired until proven otherwise.[5]
- Acquired Chiari can have one or more possible pathological co-factors; any of which can result in the descent of the cerebellar tonsils. Many Chiarians often mistakenly conceptualize an Acquired Chiari Malformation as being brought on only by trauma; however, “acquired” is an antonym for “congenital,” so an Acquired Chiari Malformation in medical terms is one that a person was not born with. While this can include Acquired Chiari malformations resulting from trauma, it can also include Acquired Chiari malformations resulting from a variety of other medical conditions:
- Heritable Disorders of Connective Tissue (HDCTs), most commonly Ehlers-Danlos Syndromes (EDS), make the tonsils more prone to prolapse below the foramen magnum.
- Multiple conditions are known to create a pushing/pulling effect that can result in a tonsillar herniation. These conditions include: Intracranial Hypertension (IH), Atlantoaxial Instability and Craniocervical Instability (AAI/CCI), Tethered Cord Syndrome (TCS), and Intracranial Hypotension (cerebrospinal fluid leaks), Hydrocephalus, and a variety of cysts and brain tumors.[2]
Special care should be taken when any of these co-morbid conditions exist in conjunction with a Chiari Malformation. Before the consideration of decompression surgery, a plan should be developed which addresses each possible comorbid condition before decompression. This can reduce the likelihood of complications and/or a failed decompression surgery.
SEVEN TYPES OF CHIARI MALFORMATIONS WORTH DISCUSSING (asterisks “*” indicate commonly known types)
Chiari Zero: The lower part of the cerebellum (the cerebral tonsils) are blocking the foramen magnum, but are not descended through. Because of the cerebellum’s position, it blocks the flow of cerebrospinal fluid and all the effects of that blockage are comparable to Type 1.
Diagnosis Requirements: Symptomology; MRI showing no herniation but the low-lying tonsils that are pressing against the top of the foramen magnum; MRI showing a syrinx (despite the name, Chiari Zero is classified under Syringomyelia and not Chiari Malformation – so a syrinx is technically required for diagnoses). [6][7]
Treatment Options: With few symptoms, non-surgical treatments might be recommended. When a syrinx is present, a decompression is often recommended before the syrinx has a chance to further develop and cause additional damage to the spine. However, even when a syrinx is present, all pathological cofactors should be explored and addressed prior to decompression surgery.
Chiari 0.5: In cases of Chiari 0.5, the lower part of the cerebellum (the cerebral tonsils) are descended through the foramen magnum, but descends < 5mm (which is the measurement that some doctors use to define Chiari). Usually labeled “tonsillar ectopia” on radiology reports, the symptoms and effects of the obstruction are generally the same as those experienced with Type 1 or Chiari 1.5.[3]
Diagnosis Requirements: Symptomology; MRI showing a herniation of < 5mm, unless already properly diagnosed with a Type 1 or Chiari 1.5; presence of a syrinx is not “required” for diagnosis, but as with Chiari Zero, it illustrates that it is causing a problem obstructing the flow of cerebrospinal fluid and may be relevant when deciding between various courses of treatment.
Treatment Options: The same as Type 1 or Chiari 1.5, respectively.

*Chiari Malformation Type 1: The most common type of Chiari Malformation, Type 1 is diagnosed when the cerebral tonsils descend below the foramen magnum. Medical professionals unfamiliar with current research surrounding Chiari Zero and Chiari 0.5 (and the symptomology surrounding the blockage of cerebrospinal fluid), believe that a tonsillar herniation of less than 5mm is simply a tonsillar ectopia and only diagnose a Chiari Malformation when the descent is > 5mm. However, the 5mm requirement is controversial, and many doctors now base their diagnoses not solely on measurements, but rather on symptomology and a combination of other factors, including cine MRI’s, a patient’s symptoms, and other relevant factors.[6] Many people with a Chiari Zero, Chiari 0.5, or Type 1 can be asymptomatic for a lifetime: one large study found that approximately 30% of those with a CM measuring between 5-10mm were asymptomatic.[8] If symptoms develop, they often present in adolescence or early adulthood. Anecdotal evidence supports the proposition that once symptoms start, the symptoms often progress rapidly until the damage is stopped surgically.
Diagnosis Requirements: Symptomology; MRI indicating at least one herniated tonsil (without the brainstem descending as well).[9]
Treatment Options: Prior to surgery any/all comorbidities should be explored and treated especially if you are found to have a normal sized posterior fossa. However, if you have classic Chiari 1 Malformation with a small posterior fossa, the risks of surgery should be weighed against the severity of symptoms and the impact that symptoms are having on the patient’s quality of life. It is often recommended to treat mild symptoms with medication, with surgical options typically reserved for cases in which symptoms cause more serious medical and quality of life problems. However, symptoms do tend to progress, and studies have shown a correlation between successful decompression surgery and the amount of time between the onset of any symptoms and surgical intervention[10]. See “Decompression Surgery” below.
Chiari 1.5: This type of CM (often referred to as a “Complex Chiari”) is often acquired as opposed to congenital. Chiari 1.5 should be the diagnosis when the tonsil(s) and all/part of the lower brainstem (the medulla oblongata) has descended past the foramen magnum. This is usually indicative of another comorbid condition pushing the brainstem downward from above or pulling downward from below.[5][11][12]
Diagnosis Requirements: symptomology; MRI indicating at least one herniated tonsil AND a downward displacement of all/part of the brainstem; without the other radiological findings associated with Type 2.
Treatment Options: Treatment options can vary significantly from patient to patient depending on the cause of the Chiari 1.5. While a variety of medical options might initially be used to treat symptoms, it is extremely important that all possible causes and/or comorbidities are thoroughly investigated and treated prior to the consideration of decompression surgery. Failure to identify and treat any such conditions can increase the likelihood of a failed decompression and further complications such as brain slumping, increased cervical instability, etc.
*Chiari Malformation Type 2 (also known as Arnold Chiari Malformation): Type 2 involves a herniation of the cerebellar tonsils and lower part of the brainstem (the medulla oblongata). Unlike in Chiari 1.5, in Type 2 the fourth ventricle is usually herniated, all/part of the cerebellar vermis (the tissue connecting both halves of the cerebellum) is missing or herniated, the corpus callosum (nerve fibers connecting both hemispheres of the brain) is fully/partially absent (agenesis), and it is almost always accompanied by a myelomeningocele (the most serious form of Spina Bifida, a congenital neural tube defect where the spinal canal does not close properly).[13][14][15]
Diagnosis Requirements: While a myelomeningocele is usually evident and diagnosed at birth, a brain MRI should confirm the radiological aspects of Type 2.
Treatment Options: Myelomeningocele is usually treated surgically at birth. If other related problems develop, such as hydrocephalus and/or tethered cord, they are often dealt with surgically as they become problematic. While some with Type 2 are decompressed, anecdotal evidence reflects a general trend of an increased failure rate with decompression surgeries as compared to those with Type 1. Because of this, some neurosurgeons choose not to decompress those with Type 2.
*Chiari Malformation Type 3: Type 3 is a serious type of Chiari Malformation involving herniated cerebellar tonsils, brainstem, and fourth ventricle. However, in most cases of Type 3, a sac forms out of the back of the skull (encephalocele) that contains brain matter from the cerebellum and the meninges. Type 3 causes severe neurological problems that are evident at birth and has a high infant mortality rate.[16][17]
*Chiari Malformation Type 4: Type 4 is the most severe type of Chiari Malformation, but does not involve a hindbrain herniation (and therefore arguments have been made that it is not a Chiari Malformation). Instead, it consists of an undeveloped or underdeveloped cerebellum. Most infants born with Type 4 die in infancy.[16][17]
SURGICAL INTERVENTION
Decompression surgery is currently the only available means of attempting to stop the progression of symptoms of a congenital chiari (with no other pathological cofactors), but decompression is not a cure (not even close). Statistics show that up to 69% of decompressed patients find some measure of relief from surgery (usually headaches)[18]. Most neurosurgeons will give only a 50% chance of helping each individual symptom. Some of the symptoms are irreversible once they develop. Recent studies show that there is a correlation between early surgical intervention and positive post-surgical outcomes.[19] However, we cannot over emphasize the importance of your doctors taking time to find, diagnose, and treat co-morbid conditions BEFORE decompression surgery. If they are not willing to consider comorbidities, they are probably not the doctor for you!
[wpedon id=”4396″ align=”center”]
*Original version released January 2018, revised October 2018.
References:
1 Eltorai, Ibrahim M. “Rare Diseases and Syndromes of the Spinal Cord” Cham: Springer International Publishing: Imprint: Springer, 2016. Page 43, 15.2, <www.springer.com/us/book/9783319451466>.
2 Henderson, Fraser C., et al. “Neurological and Spinal Manifestations of the Ehlers–Danlos Syndromes.” American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 21 Feb. 2017, <www.onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31549/full>.
3 Sekula, Raymond F, et al. “Dimensions of the Posterior Fossa in Patients Symptomatic for Chiari I Malformation but without Cerebellar Tonsillar Descent.” Cerebrospinal Fluid Research, BioMed Central, 2005, <www.ncbi.nlm.nih.gov/pmc/articles/PMC1343586>.
4 Stagi, Stefano, et al. “The Ever-Expanding Conundrum of Primary Osteoporosis: Aetiopathogenesis, Diagnosis, and Treatment.” Italian Journal of Pediatrics, BioMed Central, 2014, <www.ncbi.nlm.nih.gov/pmc/articles/PMC4064514>.
5 Milhorat, Thomas H., et al. “Mechanisms of Cerebellar Tonsil Herniation in Patients with Chiari Malformations as Guide to Clinical Management.” Acta Neurochirurgica, Springer Vienna, July 2010, <www.ncbi.nlm.nih.gov/pmc/articles/PMC2887504>.
6 Isik, N, et al. “A New Entity: Chiari Zero Malformation and Its Surgical Method.” Turkish Neurosurgery., U.S. National Library of Medicine, <www.ncbi.nlm.nih.gov/pubmed/21534216>.
7 “JNS JOURNAL OF Neurosurgery OFFICIAL JOURNALS OF THE AANS since 1944.” The Resolution of Syringohydromyelia without Hindbrain Herniation after Posterior Fossa Decompression | Journal of Neurosurgery, Vol 89, No 2, <www.thejns.org/doi/abs/10.3171/jns.1998.89.2.0212?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dpubmed>.
8 Elster, A D, and M Y Chen. “Chiari I Malformations: Clinical and Radiologic Reappraisal.”Radiology., U.S. National Library of Medicine, May 1992, <www.ncbi.nlm.nih.gov/pubmed/1561334>.
9 Wilson, Eugene. “Chiari.” CEDSA Home, <www.cedsa.org/index.php/59-quick-reference/73-chiari.html>.
10 Hindawi. “Surgical Management of Patients with Chiari I Malformation.” International Journal of Pediatrics, Hindawi, 28 June 2012, <www.hindawi.com/journals/ijpedi/2012/640127>.
11 Kim, In-Kyeong, et al. “Chiari 1.5 Malformation : An Advanced Form of Chiari I Malformation.”Journal of Korean Neurosurgical Society, The Korean Neurosurgical Society, Oct. 2010, <www.ncbi.nlm.nih.gov/pmc/articles/PMC2982921>.
12 Malik, Amita, et al. Chiari 1.5: A Lesser Known Entity. Annals of Indian Academy of Neurology, <www.annalsofian.org/article.asp?issn=0972-2327;year=2015;volume=18;issue=4;spage=449;epage=450;aulast=Malik>.
13 Wolpert, Samuel M, et al. “Chiari II Malformation: MR Imaging.” American Journal of Roentgenology, <www.ajronline.org/doi/pdf/10.2214/ajr.149.5.1033>.
14 Yumer, M H, et al. “Chiari Type II Malformation: a Case Report and Review of Literature.”Folia Medica., U.S. National Library of Medicine, <www.ncbi.nlm.nih.gov/pubmed/16918056>.
15 Kim, Irene. “Chiari II Decompression in Patients with Myelomeningocele in the National Spina Bifida Patient Registry (NSBPR).” <http://spinabifidaassociation.org/sbworldcongress/wp-content/uploads/sites/10/2017/04/B.4-Kim-Neurosurgery.pdf>.
16 “Chiari Malformation Fact Sheet.” National Institute of Neurological Disorders and Stroke, U.S. Department of Health and Human Services, <www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Chiari-Malformation-Fact-Sheet>.
17 “Chiari Malformations.” NORD (National Organization for Rare Disorders), <www.rarediseases.org/rare-diseases/chiari-malformations>.
18 14 Aliaga, L, et al. “A Novel Scoring System for Assessing Chiari Malformation Type I Treatment Outcomes.” Neurosurgery., U.S. National Library of Medicine, Mar. 2012, <www.ncbi.nlm.nih.gov/pubmed/21849925>.
19 Siasios, John, et al. “Surgical Management of Patients with Chiari I Malformation” International Journal of Pediatrics, Article ID 640127, Hindawi, 2012, <www.hindawi.com/journals/ijpedi/2012/640127>.


 But to look at the full history of what became known as a Chiari Malformation, we can begin by looking at the research of a German pathologist, named Theodor Langhans. In his research in 1881 (a decade before Hans Chiari conducted his research on what became known as a Chiari Malformation), while looking at syringomyelia (“a cavity created in the spinal cord”), he noted a “change in the cerebellar cavity.” Upon dissection of the cerebellum, he described the cerebellar tonsils as “two symmetrical pyramidal tumors,” pushing the brainstem forward.
But to look at the full history of what became known as a Chiari Malformation, we can begin by looking at the research of a German pathologist, named Theodor Langhans. In his research in 1881 (a decade before Hans Chiari conducted his research on what became known as a Chiari Malformation), while looking at syringomyelia (“a cavity created in the spinal cord”), he noted a “change in the cerebellar cavity.” Upon dissection of the cerebellum, he described the cerebellar tonsils as “two symmetrical pyramidal tumors,” pushing the brainstem forward. Unfortunately it leaves most of us with failed decompressions, fighting with our neurosurgeons that “something is still wrong.” These neurosurgeons look at their post-operative checklist and see that they successfully did everything surgically required in their out-of-date textbooks:
Unfortunately it leaves most of us with failed decompressions, fighting with our neurosurgeons that “something is still wrong.” These neurosurgeons look at their post-operative checklist and see that they successfully did everything surgically required in their out-of-date textbooks:



 I was what you would consider a “typical developing child” growing up, I did not have any health issues and was able to enjoy much of my childhood. My journey to finding answers in regard my health began at 15 years old, when I began rapidly losing my vision in my left eye. I dealt with severe headaches and the doctors struggled to draw a connection to my declining vision. I went from 20/20 vision to 20/400 in my right eye and 0 vision in my left. I was considered legally blind. I had to relearn how to navigate life with very little vision.
I was what you would consider a “typical developing child” growing up, I did not have any health issues and was able to enjoy much of my childhood. My journey to finding answers in regard my health began at 15 years old, when I began rapidly losing my vision in my left eye. I dealt with severe headaches and the doctors struggled to draw a connection to my declining vision. I went from 20/20 vision to 20/400 in my right eye and 0 vision in my left. I was considered legally blind. I had to relearn how to navigate life with very little vision. Fast forward a few years, I was told that a second decompression surgery was required, which I agreed to. It resulted in a rip in my dural patch causing a cerebral spinal fluid leak at the surgical site. After these two decompression and a CSF leak repair surgery, my vision had improved significantly yet I was worse off symptom wise than I was when I initially began noticing changes in my body. Sadly, I was told from my specialists that there was nothing more they could do for me. They referred me to the headache/face pain clinic. After many failed attempts at managing my pain with medications, my doctor mentioned that my symptoms resembled a spinal fluid leak and that there is a doctor who is navigating research and I should be evaluated.
Fast forward a few years, I was told that a second decompression surgery was required, which I agreed to. It resulted in a rip in my dural patch causing a cerebral spinal fluid leak at the surgical site. After these two decompression and a CSF leak repair surgery, my vision had improved significantly yet I was worse off symptom wise than I was when I initially began noticing changes in my body. Sadly, I was told from my specialists that there was nothing more they could do for me. They referred me to the headache/face pain clinic. After many failed attempts at managing my pain with medications, my doctor mentioned that my symptoms resembled a spinal fluid leak and that there is a doctor who is navigating research and I should be evaluated. contrast, CT , digital subtraction myelogram, MR myelogram, and the list goes on. After a few months of investigating we were able to confirm that I suffer from spontaneous intracranial hypotension, meaning that I have multiple leaks or suspicious areas in my spine, that happened spontaneously (without known trauma). My doctor mentioned that my Chiari diagnosis is what is classified as an Acquired Chiari Malformation.
contrast, CT , digital subtraction myelogram, MR myelogram, and the list goes on. After a few months of investigating we were able to confirm that I suffer from spontaneous intracranial hypotension, meaning that I have multiple leaks or suspicious areas in my spine, that happened spontaneously (without known trauma). My doctor mentioned that my Chiari diagnosis is what is classified as an Acquired Chiari Malformation.

 After several hours I regained consciousness, family and doctors surrounded me trying to explain what they knew about baby Eric. They explained Eric and I would have to be immediately flown out to a larger center where they could properly care for my baby. Eric couldn’t support his own airway, he couldn’t suck, swallow, or move due to hypertonic muscles. He was having several seizures, all pointed to brain damage. At this point they had no idea what was wrong and could not conclude whether my baby would live. I recall saying I wanted to go see my baby and trying to get up out of the hospital bed, but the nurses told me I couldn’t see him, because I had just gone through a major surgery and he was too sick; we both had to rest for transport. When the nurses told me that, I flipped, telling them that come hell or high water, I was going to see him. I got as far as swinging my legs over the bed before I vomited everywhere. The nurses finally clued in that I was going with or without their help, so they laid me back down and wheeled me to the NICU to see him. I couldn’t even see my baby for he was so small and leads encased his body where he layed. They had his chin strapped up as every time it fell backward it would close off his airway leading to oxygen desaturation. They didn’t have the resources to intubate a baby at this hospital, so they decided to fly us out to Vancouver Children’s Hospital.
After several hours I regained consciousness, family and doctors surrounded me trying to explain what they knew about baby Eric. They explained Eric and I would have to be immediately flown out to a larger center where they could properly care for my baby. Eric couldn’t support his own airway, he couldn’t suck, swallow, or move due to hypertonic muscles. He was having several seizures, all pointed to brain damage. At this point they had no idea what was wrong and could not conclude whether my baby would live. I recall saying I wanted to go see my baby and trying to get up out of the hospital bed, but the nurses told me I couldn’t see him, because I had just gone through a major surgery and he was too sick; we both had to rest for transport. When the nurses told me that, I flipped, telling them that come hell or high water, I was going to see him. I got as far as swinging my legs over the bed before I vomited everywhere. The nurses finally clued in that I was going with or without their help, so they laid me back down and wheeled me to the NICU to see him. I couldn’t even see my baby for he was so small and leads encased his body where he layed. They had his chin strapped up as every time it fell backward it would close off his airway leading to oxygen desaturation. They didn’t have the resources to intubate a baby at this hospital, so they decided to fly us out to Vancouver Children’s Hospital. begging, pleading to God for your child to be healed. After 3 weeks they finally let me have skin to skin cuddles and started teaching us how to care for him. This isn’t the way it’s supposed to go; you’re supposed to go to the hospital, have a baby and take them home, right? Instead you walk around numb, you don’t remember the last time you ate, or showered, you just feel like you’re having an out of body experience, as if you’re watching this happen to someone else. You’re scared to leave their side for even a minute because what if it’s that minute that he leaves this world and you’re not there?
begging, pleading to God for your child to be healed. After 3 weeks they finally let me have skin to skin cuddles and started teaching us how to care for him. This isn’t the way it’s supposed to go; you’re supposed to go to the hospital, have a baby and take them home, right? Instead you walk around numb, you don’t remember the last time you ate, or showered, you just feel like you’re having an out of body experience, as if you’re watching this happen to someone else. You’re scared to leave their side for even a minute because what if it’s that minute that he leaves this world and you’re not there? After about a month we had a meeting, and it was then that the doctors told us we had a failure to thrive baby, his cells didn’t migrate to the right place at the right time, that his cerebellum and brainstem were severely underdeveloped. The statistics 19 years ago was that one out of one million babies were born like this and they had yet to find out what caused the illness. Our baby couldn’t regrow the parts of his brain that didn’t develop. He had gestational arrest at 32 weeks, he had scoliosis, spina bifida occulta, epilepsy and severe brain damage. Our child would never move on his own, suck or swallow or be able to interact and that he would most likely die of aspiration pneumonia. He would live his life in hospital more than out. I have worked with special needs adults with this type of quality of life, I could play the tape to the end, and I knew hanging on to him would only be for selfish reasons. I understood that my baby was in pain, I could see in his beautiful blue eyes, as I said before, a mother always knows. It was at this time Eric’s father and I decided to sign a” Do Not Resuscitate” order. It was without a doubt the single hardest thing I have ever done.
After about a month we had a meeting, and it was then that the doctors told us we had a failure to thrive baby, his cells didn’t migrate to the right place at the right time, that his cerebellum and brainstem were severely underdeveloped. The statistics 19 years ago was that one out of one million babies were born like this and they had yet to find out what caused the illness. Our baby couldn’t regrow the parts of his brain that didn’t develop. He had gestational arrest at 32 weeks, he had scoliosis, spina bifida occulta, epilepsy and severe brain damage. Our child would never move on his own, suck or swallow or be able to interact and that he would most likely die of aspiration pneumonia. He would live his life in hospital more than out. I have worked with special needs adults with this type of quality of life, I could play the tape to the end, and I knew hanging on to him would only be for selfish reasons. I understood that my baby was in pain, I could see in his beautiful blue eyes, as I said before, a mother always knows. It was at this time Eric’s father and I decided to sign a” Do Not Resuscitate” order. It was without a doubt the single hardest thing I have ever done. Within 2 weeks, it was clear this child was a strong fighter and wasn’t ready to give up quite yet. We had another meeting and it was decided we would take him home. We wanted his big sister to have time with him and show him what a home was like. We took a 2-week crash course on neonatal nursing. We had to learn how to do his lung physio, how to suction him, and how to work a feeding pump and so much more. Eric was brought home February 10,1999. We did his 24-hour care until March 9th when he took a turn for the worse. He was diagnosed with aspiration pneumonia. This left me in a panic; I wasn’t ready to let him go, and I wanted him sent back to Children’s Hospital and be treated. We had an amazing Doctor who came to the hospital and took me for a walk to discuss why we made the DNR code and why we made the right choice for our son. I took my son home that day knowing we were running on borrowed time. His breathing became very shallow, he turned blue from lack of oxygen and on March 11, 1999 at 3:15 am our darling boy went home to be with his creator. The year following is a fog, I
Within 2 weeks, it was clear this child was a strong fighter and wasn’t ready to give up quite yet. We had another meeting and it was decided we would take him home. We wanted his big sister to have time with him and show him what a home was like. We took a 2-week crash course on neonatal nursing. We had to learn how to do his lung physio, how to suction him, and how to work a feeding pump and so much more. Eric was brought home February 10,1999. We did his 24-hour care until March 9th when he took a turn for the worse. He was diagnosed with aspiration pneumonia. This left me in a panic; I wasn’t ready to let him go, and I wanted him sent back to Children’s Hospital and be treated. We had an amazing Doctor who came to the hospital and took me for a walk to discuss why we made the DNR code and why we made the right choice for our son. I took my son home that day knowing we were running on borrowed time. His breathing became very shallow, he turned blue from lack of oxygen and on March 11, 1999 at 3:15 am our darling boy went home to be with his creator. The year following is a fog, I  remember very little. I was deeply depressed but I knew I had to keep moving forward for my daughter, she needed me. I know she was hurting too but I was so consumed with my own grief, that I couldn’t reach out to her, I couldn’t handle both her grief and mine. My daughter and I have had to take a lot of time since to heal together. With that being said, the pain of losing a child is not something that you can run away from or attempt to forget; I relive my sons small time on this earth every single day.
remember very little. I was deeply depressed but I knew I had to keep moving forward for my daughter, she needed me. I know she was hurting too but I was so consumed with my own grief, that I couldn’t reach out to her, I couldn’t handle both her grief and mine. My daughter and I have had to take a lot of time since to heal together. With that being said, the pain of losing a child is not something that you can run away from or attempt to forget; I relive my sons small time on this earth every single day.

 I had a rough journey with these conditions and I am sharing it to help show how complex we can be and how much we need the medical community to step it up a notch (or ten)! I grew up in Denmark, where I lived when I was diagnosed and had my first surgeries.
I had a rough journey with these conditions and I am sharing it to help show how complex we can be and how much we need the medical community to step it up a notch (or ten)! I grew up in Denmark, where I lived when I was diagnosed and had my first surgeries.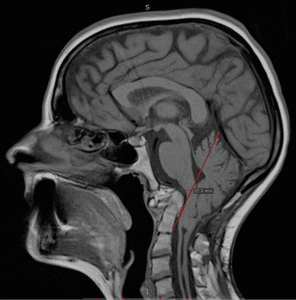 Years passed by with various periods of severe pain flares, flares that felt nothing like the pains I had before. But no doctor was really believing me. I had a spontaneous collapsed lung a couple of times in this period and ended up with surgery for this (I woke up under this surgery which later also turned out to have a significance). At 25, one day I had a sudden and severe onset of symptoms – a pain in the back of my head feeling like two stabbing knives. This did not resolve and after several attempts with various medicines, that I only got sicker from, I finally saw a new rheumatologist whom again treated me with harsh accusations of laziness and psychological imbalance. I can assure you he was the one bringing on my tears that day, despite the extra severe pain I had been in for weeks. I was placed in the care of the hospital physios and after a while, it became clear to them that there was something really wrong and they got me to see another rheumatologist, who in turn took their word and referred me for an MRI. I had only just turned 26 when I was diagnosed with Chiari 1 Malformation and Syringomyelia – in my full spine.
Years passed by with various periods of severe pain flares, flares that felt nothing like the pains I had before. But no doctor was really believing me. I had a spontaneous collapsed lung a couple of times in this period and ended up with surgery for this (I woke up under this surgery which later also turned out to have a significance). At 25, one day I had a sudden and severe onset of symptoms – a pain in the back of my head feeling like two stabbing knives. This did not resolve and after several attempts with various medicines, that I only got sicker from, I finally saw a new rheumatologist whom again treated me with harsh accusations of laziness and psychological imbalance. I can assure you he was the one bringing on my tears that day, despite the extra severe pain I had been in for weeks. I was placed in the care of the hospital physios and after a while, it became clear to them that there was something really wrong and they got me to see another rheumatologist, who in turn took their word and referred me for an MRI. I had only just turned 26 when I was diagnosed with Chiari 1 Malformation and Syringomyelia – in my full spine.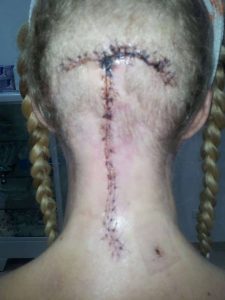 I did a bit of internet research as information in Danish was very sparse and realized there was a certain number of bad outcomes due to something called Ehlers-Danlos Syndrome, Retroflexed Odontoid and Basilar Invagination. I asked my neurosurgeon, who was supposed to be the best in Denmark at this point, about these things – he claimed I didn’t have any. However, he did agree to refer me for Ehlers-Danlos evaluation. Here I was told I did not have that either. However, I was bordering on a similar connective tissue disorder called Marfan Syndrome, which they still could not diagnose me with due to my heart and eyes not being faulty. I had my first decompression surgery in December 2006. It was rough. I reacted badly to anesthetics and to the morphine and I also lost a lot of spinal fluid. I could not raise myself up the first month which I thought was normal. Slowly, I got better, and imaging showed my syrinx shrinking. Two and a half years later, though, I started experiencing dizziness and nausea and though my first surgeon didn’t believe me, imaging finally showed a big collection of fluid outside my spinal cord originating from a hole in the duraplasty used to close after tonsillar cauterization at my first surgery. I tried talking to the surgeon about concerns of Klippel-Feil and instability, that I had read about, but they would not hear of it and said that for now they would just focus on this issue. So, this was repaired, and I moved to Spain with my boyfriend at the time. I was placed on a disability pension from Denmark and that enrolled me in the Spanish public healthcare. I did, however, in the meantime follow up on my concerns and contacted a specialist, who had written about the Klippel-Feil and Chiari connection, and he straight away stated I had some severe issues with my odontoid and needed it removed and my neck fused to my skull. My first meeting with a surgeon in Spanish health care came up and he just looked at my imaging two minutes then stated my problems were way bigger than Chiari and Syringomyelia. He also diagnosed a severe retroflexed odontoid and Basilar Invagination – so severe he had a hard time understanding how I could breathe, let alone walk. But given my reasonably good condition, he opted to postpone these surgeries as they are big and not without risks.
I did a bit of internet research as information in Danish was very sparse and realized there was a certain number of bad outcomes due to something called Ehlers-Danlos Syndrome, Retroflexed Odontoid and Basilar Invagination. I asked my neurosurgeon, who was supposed to be the best in Denmark at this point, about these things – he claimed I didn’t have any. However, he did agree to refer me for Ehlers-Danlos evaluation. Here I was told I did not have that either. However, I was bordering on a similar connective tissue disorder called Marfan Syndrome, which they still could not diagnose me with due to my heart and eyes not being faulty. I had my first decompression surgery in December 2006. It was rough. I reacted badly to anesthetics and to the morphine and I also lost a lot of spinal fluid. I could not raise myself up the first month which I thought was normal. Slowly, I got better, and imaging showed my syrinx shrinking. Two and a half years later, though, I started experiencing dizziness and nausea and though my first surgeon didn’t believe me, imaging finally showed a big collection of fluid outside my spinal cord originating from a hole in the duraplasty used to close after tonsillar cauterization at my first surgery. I tried talking to the surgeon about concerns of Klippel-Feil and instability, that I had read about, but they would not hear of it and said that for now they would just focus on this issue. So, this was repaired, and I moved to Spain with my boyfriend at the time. I was placed on a disability pension from Denmark and that enrolled me in the Spanish public healthcare. I did, however, in the meantime follow up on my concerns and contacted a specialist, who had written about the Klippel-Feil and Chiari connection, and he straight away stated I had some severe issues with my odontoid and needed it removed and my neck fused to my skull. My first meeting with a surgeon in Spanish health care came up and he just looked at my imaging two minutes then stated my problems were way bigger than Chiari and Syringomyelia. He also diagnosed a severe retroflexed odontoid and Basilar Invagination – so severe he had a hard time understanding how I could breathe, let alone walk. But given my reasonably good condition, he opted to postpone these surgeries as they are big and not without risks. A couple of years of enjoying the benefits the climate change gave me (and likely putting my head in the sand) went by but then I could no longer ignore the fact that I was getting worse. I was in a rough period with other matters of life, so it took a while before I realized I couldn’t escape the changes in my body. I started losing weight amongst other things and after a quick detour of fear of stomach cancer, I finally realized that everything that was going on was related to my brainstem compression. So, I went back to the neurosurgeon. He ordered some testing but before it could be done, I ended up admitted urgently after I stopped breathing one night. From here started a roller coaster. I didn’t feel right about their suggestions and the surgeon that was going to operate didn’t feel very secure himself even. I ended up getting transferred to a private hospital in Barcelona that calls themselves a “Chiari Institute.” Had I known what I do now, I would never have paid the fee for a filum release, but the doctor claimed this was what I needed and well… It was worth a shot in this urgent situation. He then sent me home, claiming I was cured. I didn’t feel right and breathing through the night was still a problem, so I started sending my imaging to experts around the world and working on getting referred to another hospital in Spain’s public health with higher expertise. All these experts claimed I wouldn’t have long to live unless I had this odontoid approached.
A couple of years of enjoying the benefits the climate change gave me (and likely putting my head in the sand) went by but then I could no longer ignore the fact that I was getting worse. I was in a rough period with other matters of life, so it took a while before I realized I couldn’t escape the changes in my body. I started losing weight amongst other things and after a quick detour of fear of stomach cancer, I finally realized that everything that was going on was related to my brainstem compression. So, I went back to the neurosurgeon. He ordered some testing but before it could be done, I ended up admitted urgently after I stopped breathing one night. From here started a roller coaster. I didn’t feel right about their suggestions and the surgeon that was going to operate didn’t feel very secure himself even. I ended up getting transferred to a private hospital in Barcelona that calls themselves a “Chiari Institute.” Had I known what I do now, I would never have paid the fee for a filum release, but the doctor claimed this was what I needed and well… It was worth a shot in this urgent situation. He then sent me home, claiming I was cured. I didn’t feel right and breathing through the night was still a problem, so I started sending my imaging to experts around the world and working on getting referred to another hospital in Spain’s public health with higher expertise. All these experts claimed I wouldn’t have long to live unless I had this odontoid approached. October 2016 I finally had a partial odontoidectomy and a fusion, which beyond doubt saved my life. It was a rough ride, for both me and the surgeons. They had to deal with complications related to my anatomy, to the mess the first surgeon in Denmark had left – he had indeed damaged my muscles more than I ever knew – and to the problems relating to the soft tissue. I do know they did a great job, but due to all the mistakes, how complicated my case was and is, I am unfortunately not done. I have ongoing issues and though some of these could have been avoided with the right approach from the beginning, some are just the way it is with these conditions.
October 2016 I finally had a partial odontoidectomy and a fusion, which beyond doubt saved my life. It was a rough ride, for both me and the surgeons. They had to deal with complications related to my anatomy, to the mess the first surgeon in Denmark had left – he had indeed damaged my muscles more than I ever knew – and to the problems relating to the soft tissue. I do know they did a great job, but due to all the mistakes, how complicated my case was and is, I am unfortunately not done. I have ongoing issues and though some of these could have been avoided with the right approach from the beginning, some are just the way it is with these conditions. Have you written something that you think would be of value to the Chiari community? Consider publishing it with us! It might be exactly what someone else needs to hear for them to make it through their next mile of the fight!
Have you written something that you think would be of value to the Chiari community? Consider publishing it with us! It might be exactly what someone else needs to hear for them to make it through their next mile of the fight!
 INCURABLE DISEASES. THE GENERAL ASSEMBLY OF THE UNITED NATIONS HAS GONE ON RECORD STATING THAT, “UNTREATED PAIN IS TANTAMOUNT TO TORTURE OR CRUEL, INHUMAN OR DEGRADING TREATMENT OR PUNISHMENT.”
INCURABLE DISEASES. THE GENERAL ASSEMBLY OF THE UNITED NATIONS HAS GONE ON RECORD STATING THAT, “UNTREATED PAIN IS TANTAMOUNT TO TORTURE OR CRUEL, INHUMAN OR DEGRADING TREATMENT OR PUNISHMENT.”  “Opioid Crisis” is a new buzz term being overused by national news and government officials to evoke a negative feeling about a drug that was created for helping people. The definition of “crisis” by Merriam-Webster dictionary is partially defined as “an unstable or crucial time or state of affairs in which a decisive change is impending; especially: one with the distinct possibility of a highly undesirable outcome.”
“Opioid Crisis” is a new buzz term being overused by national news and government officials to evoke a negative feeling about a drug that was created for helping people. The definition of “crisis” by Merriam-Webster dictionary is partially defined as “an unstable or crucial time or state of affairs in which a decisive change is impending; especially: one with the distinct possibility of a highly undesirable outcome.” There is also a negative stigma attached to having to take pain meds. Many chronic pain patients take prescription opioids to continue meaningful work. This could be a huge problem because they are afraid to speak out, afraid of losing their job, or at least bringing suspicion upon themselves from employers. Many patients are afraid that if those around them know they take pain medication, they will be seen as addicts. But it is time to brush off the stigma and the fear and to stand up for our rights to access proper and effective medical treatment. If we don’t, we may lose access permanently and keeping our jobs, social lives, and other important activities and relationships will not be possible due to disability from pain.
There is also a negative stigma attached to having to take pain meds. Many chronic pain patients take prescription opioids to continue meaningful work. This could be a huge problem because they are afraid to speak out, afraid of losing their job, or at least bringing suspicion upon themselves from employers. Many patients are afraid that if those around them know they take pain medication, they will be seen as addicts. But it is time to brush off the stigma and the fear and to stand up for our rights to access proper and effective medical treatment. If we don’t, we may lose access permanently and keeping our jobs, social lives, and other important activities and relationships will not be possible due to disability from pain. Doctors that want to be doctors are now caught up wasting time in government “guidelines” or laws and pressure from insurance companies that do not want to cover pain control prescribed by our doctors. Many doctors who have continued to prescribe despite the current hostile regulatory environment have been threatened, raided, and even arrested by the DEA.
Doctors that want to be doctors are now caught up wasting time in government “guidelines” or laws and pressure from insurance companies that do not want to cover pain control prescribed by our doctors. Many doctors who have continued to prescribe despite the current hostile regulatory environment have been threatened, raided, and even arrested by the DEA.
 Empowering With Knowledge!
Empowering With Knowledge!  be, but if we continue to see all that we can do and not just what we cannot do, we can dare to dream again! We are all multifaceted human beings with broad gifts and talents. We might not be the athletes we once aspired to be, but that says nothing of our strength. We all have the potential to change the world around us! You might be an artist that hasn’t practiced your art in years – start practicing again! You might have always thought about writing books, but because of your diminished hope for the future, you haven’t written in years – pick up a pen and start writing again! The only way to ever know your true potential is to try and try again! Dare to dream again!
be, but if we continue to see all that we can do and not just what we cannot do, we can dare to dream again! We are all multifaceted human beings with broad gifts and talents. We might not be the athletes we once aspired to be, but that says nothing of our strength. We all have the potential to change the world around us! You might be an artist that hasn’t practiced your art in years – start practicing again! You might have always thought about writing books, but because of your diminished hope for the future, you haven’t written in years – pick up a pen and start writing again! The only way to ever know your true potential is to try and try again! Dare to dream again!